“Well, in our country,” said Alice, still panting a little, “you’d generally get to somewhere else — if you run very fast for a long time, as we’ve been doing.”
“A slow sort of country!” said the Queen. “Now, here, you see, it takes all the running you can do, to keep in the same place. If you want to get somewhere else, you must run at least twice as fast as that!”
Through the Looking Glass and what Alice Found There Lewis Carroll
The Red Queen hypothesis is well-accepted in evolutionary biology. Organisms evolve and adapt not to gain an evolutionary advantage, but simply to not fall behind competing organisms that evolve and adapt. Hence, everyone has to “run as fast as they can” (evolve) to “stay in the same place” (reproduce). It’s a nice hypothesis, and has been shown to be fairly descriptive when dealing with close competitors, such as host-parasite or predator-prey relationships.
Which is why the title of this paper published in mBio has piqued my interest: “The Black Queen Hypothesis: Evolution of Dependencies through Adaptive Gene Loss”. What is the Black Queen hypothesis?
The Red Queen in Alice was a chess piece. (And not, as the authors say in the paper, a card). The Black Queen is from a card game: namely, the Queen of Spades in the game of Hearts. Hearts is a three to five player card game, and the idea is to avoid taking tricks containing certain cards. Anything in hearts suite is bad, with one penalty point per card. But the Queen of Spades is particularly horrible, with 13 penalty points. Thus, the idea is to avoid taking hearts or the Queen of Spades.
Are you kidding? I spent three weeks at Camp Winiwinaia on Lake George the summer I was twelve. YMCA camp — poor kids’ camp my mother called it. It rained practically every day, and all we did was play Hearts and hunt The Bitch.
Hearts in Atlantis Stephen King
The authors of the paper use Hearts to set a model explaining reductive evolution in bacteria. Why would some bacterial lineages of free-living bacteria lose genes? How does an evasion trick card game tie into evolution?
In the context of evolution, the BQH (Black Queen Hypothesis IF) posits that certain genes, or more broadly, biological functions, are analogous to the queen of spades. Such functions are costly and therefore undesirable, leading to a selective advantage for organisms that stop performing them. At the same time, the function must provide an indispensable public good, necessitating its retention by at least a subset of the individuals in the community—after all, one cannot play Hearts without a queen of spades.
One such Black Queen card is the catalase-peroxidase gene, katG. katG protects against hydrogen peroxide (H2O2), a toxic byproduct of marine photosynthesis. The catalase-peroxidase protein is iron dependent, and its synthesis can be expensive, especially in an iron-poor environment. Two common marine cyanobacteria are Synenchococcus and Prochlorococcus, which typically are found in the same communities. Most Prochlorococcus lack the katG gene in their genome, while Synechococcus do have it. It seems that in ocean-surface communities, Synechococcus is holding the katG Black Queen gene in the game, while Procholorococcus elegantly avoided taking that costly card. Synechococcus is the workhorse of reducing the toxic H2O2, while the katG-deficient bacteria enjoy the common benefits to all. So it is best to be a member of a lineage that avoids having katG, while living in close proximity to the lineages that have katG. The figure below shows that the entire Prochlorococcus clade (green) lacks katG, but (presumably), living in a community with Synechococcus, allows it to benefit from the katG gene carried by the latter.
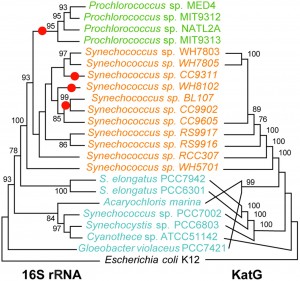
Comparison between the phylogenies of the catalase-peroxidase and small subunit rRNA genes for cyanobacteria with sequenced genomes. Although there are some differences in branching order between the two trees, the marine Synechococcus KatG proteins form a well-supported monophyletic clade, implying that this protein was present in the clade’s ancestor and was subsequently lost in several lineages (indicated by red dots on the rRNA tree), including Prochlorococcus. Green, representatives of the Prochlorococcus clade; orange, marine Synechococcus clade; cyan, other Cyanobacteria. Bootstrap values less than 75% are omitted. Only the tree topologies are shown; branch lengths do not represent genetic distances.
Leaky Functions
The authors talk about “leaky functions”: functions that provide advantage to the community in a way that is unintentionally altruistic: if an organism has the ability to extracellularly protect against H2O2, and that species lived in a community, others will benefit. However, the BQH model predicts that lineages will continue to lose leaky functions, as long as at least one lineage maintains it, benefiting the community. Should that lineage lose the leaky function, or be removed form the community, the effects could be devastating to the community now lacking that leaky function. In other words, leaky-function species eventually become keystone species of their ecosystem.
My two cents worth: I like the model. Like any good model, it provides us with testable hypotheses, and if it works well it will provide predictive powers to evolutionary changes in microbial communities. It can explain the rarity of some essential functions in a microbial community, and possibly why so many microbes fail to grow in pure culture. Time will tell how well this model will work. My only problem is that I am not sure I agree with the title the authors gave their model. Getting the Queen of Spades in Hearts is devastating to your hand (you basically lose). That would be the genetic equivalent of a cell going apoptotic (killing itself) following a cancer mutation or a viral infection. The BQH model is more subtle, an evolutionary cost-benefit model via the leaky function mechanism. Maybe the “volunteer fire-brigade hypothesis”? Or a generic: “it’s a dirty job but somebody’s got to do it” hypothesis?
Morris, J., Lenski, R., & Zinser, E. (2012). The Black Queen Hypothesis: Evolution of Dependencies through Adaptive Gene Loss mBio, 3 (2) DOI: 10.1128/mBio.00036-12