
Three figures from the undergrad who is always high
Seven tables from the lab tech with his heart of stone
Nine supplements from the postdocs, with careers doomed to die
One manuscript for the Editor on his dark throne
In the journal submission form, where the shadows lie
One paper to rule them all, one paper to find them
One paper to bring them in and in the darkness bind them
In the submission form, where the shadows lie













One of the major unsolved problems in bioinformatics is the protein folding problem: given an amino acid sequence, predict the overall three-dimensional structure of the corresponding protein. It has been known since the seminal work of Christian B. Anfinsen in the early seventies that the sequence of a protein encodes its structure, but the exact details of the encoding still remain elusive. Since the protein folding problem is of enormous practical, theoretical and medical importance – and in addition forms a fascinating intellectual challenge – it is often called the holy grail of bioinformatics.Currently, most protein structure prediction methods are based on rather ad hoc approaches. The aim of this project is to develop and implement a statistically rigorous method to predict the structure of proteins, building on various probabilistic models of protein structure developed by the Hamelryck group (see Bibliography). The method will also take the dynamic nature of proteins into account.
Bibliography:
Boomsma, W., Mardia, KV., Taylor, CC., Ferkinghoff-Borg, J., Krogh, A. and Hamelryck, T. (2008) A generative, probabilistic model of local protein structure. Proc. Natl. Acad. Sci. USA, 105, 8932-8937
Mardia, KV., Kent, JT., Zhang, Z., Taylor, C., Hamelryck, T. (2012) Mixtures of concentrated multivariate sine distributions with applications to bioinformatics. J. Appl. Stat. 39, 2475-2492.
Boomsma, W., Frellsen, J., Harder, T., Bottaro, S., Johansson, KE., Tian, P., Stovgaard, K., Andreetta, C., Olsson, S., Valentin, J., Antonov, L., Christensen, A., Borg, M., Jensen, J., Lindorff-Larsen, K., Ferkinghoff-Borg, J., Hamelryck, T. (2013) PHAISTOS: A framework for Markov chain Monte Carlo simulation and inference of protein structure. J. Comput. Chem. 34, 1697-705
Hamelryck, T., Mardia, KV., Ferkinghoff-Borg, J., Editors. (2012) Bayesian methods in structural bioinformatics. Book in the Springer series “Statistics for biology and health”, 385 pages, 13 chapters. Springer Verlag, March, 2012
Valentin, J., Andreetta, C., Boomsma, W., Bottaro, S., Ferkinghoff-Borg, J., Frellsen, J., Mardia, KV, Tian, P., Hamelryck, T. (2013) Formulation of probabilistic models of protein structure in atomic detail using the reference ratio method. Proteins. Accepted.
Requirements: Knowledge of statistics, machine learning and programming (C++ or equivalent). Knowledge of biology or biophysics is a plus but not a requirement.
Place of enrollment: Department of Biology, Bioinformatics Center
Supervisor: Assoc. Prof. Thomas Hamelryck
Co-supervisor: Prof. Michael Sørensen from Department of Mathematical Sciences
Apply here: http://dsin.ku.dk/positions/
We have Palaeolithic emotions, medieval institutions and God-like technologies.
E.O. Wilson
Whole genome sequencing will soon be cheap enough to be widely affordable. We are nearing the time when omics data may be retained for patients on a wide basis. These may include full exome, haplotype, full genome sequencing, tissue level transcriptomic data, microbiome and meta-transcrimtome, metabolome… the sky, or rather personal healthcare budget, is the limit.
Personal genomic advocates today present sequencing as a personal choice. When faced with concerns such as privacy, the general response from personalized medicine advocates is that the benefits outweigh privacy concerns, and in any case the people making the choice to sequence their genomes (at least now), are making an informed personal choice. This means that whatever possible detriment that may ensue from sequencing the genome will only affect that person. So, for example, denial of life insurance due to a genomic findings (legal in many countries) will only affect the person having their genome sequenced. In the US, where healthcare is privatized, denial of health insurance due to genetics is illegal, but the application of higher premiums is a concern. For example, some health insurance providers in the US charge higher premiums if the insured has two X chromosomes, although you usually don’t need full-genome sequencing to determine that genotype. Other privacy concerns may include the leaking (via legal or illegal means) of genomic information to various entities you may not want to have your DNA data.
Trouble is, getting your genome sequenced is not solely a personal choice: a person’s genomic information contains that of their family as well. So by having your own ‘omic information stored, you are making a choice for your siblings, parents, and children (including those yet unborn). So you are making a choice for them to know, or at least suspect, that they have certain genotypes they may or may not wish to know about. Michael Snyder, one of the strongest advocates of personal genomics has a habit of saying: “don’t sequence your genome if you are a worrier”. You may not be, but your unborn daughter may be. You may be able to correctly interpret the probabilistic data your genome provides, but your son or brother may not.
Or they may just be a private persons who would not want their genomic information out there, even by proxy. In realistic terms, by cross-referencing familial and genomic databases, your daughter may be denied certain health or life insurance coverage, based on a genotype an insurance company does not like: which may simply mean an over-interpretation of the limited predictive power of genomic data. By having your data accessible, some of her data are accessible as well, indirectly. No database is crack-proof, and re-identifying supposedly anonymous genomic data is surprisingly easy . Familial DNA matching, coupled with surreptitious collection of DNA is becoming common practice with law enforcement to generate suspect lists. As the availability of genomic data increases, so does the erosion of personal privacy.
This all sounds rather alarmist, counter-progressive, and may give me the appearance of a bit of a Luddite. Especially when coming from a genome scientist… What about the huge benefits that await us from personal genomics? Should privacy and unfounded (or well-founded) anxieties stand in the way of progress? My prediction: they probably won’t. As the cost of personal genomics decrease, and the benefits (currently somewhat hyped) increase, genotyping may start to be mandated by healthcare providers, and perhaps even some employers. But revisit the motto of this post: should we not, at least, consider some of the implications of our choices upon others, if not ourselves, given our “paleolithic emotions and medieval institutions”?

Source: CDC (Public Domain) http://blogs.cdc.gov/genomics/2011/08/25/think-before-you-spit-do-personal-genomic-tests-improve-health/
Because.. Django reinhardt.
Since this is Open Access Week, I thought I’d do the Open-Access / CC thing and share someone else’s work. In this case, a highly topical short story written by Richard Stallman. The author also has a constantly updated page with comments on the restrictions placed today on sharing reading materials. As you will see, this story may be not too far-fetched as it first seems…
The Right to Read
The following article appeared in the February 1997 issue of Communications of the ACM (Volume 40, Number 2).
Copyright © 1996, 2002, 2007, 2009, 2010 Richard Stallman
Reproduced under CC-BY-Noderiv license
From The Road To Tycho, a collection of articles about the antecedents of the Lunarian Revolution, published in Luna City in 2096.
For Dan Halbert, the road to Tycho began in college—when Lissa Lenz asked to borrow his computer. Hers had broken down, and unless she could borrow another, she would fail her midterm project. There was no one she dared ask, except Dan.
This put Dan in a dilemma. He had to help her—but if he lent her his computer, she might read his books. Aside from the fact that you could go to prison for many years for letting someone else read your books, the very idea shocked him at first. Like everyone, he had been taught since elementary school that sharing books was nasty and wrong—something that only pirates would do.
Continue reading The Right to Read →
Hi all,
I’m happy to say that the 2014 International Biocuration Conference is off to a good start. I have attended this excellent meeting twice before, and this year I am honored to be on the organizing committee. There was a lot of work behind the scenes, and we have agreed on five session topics. I am co-chairing the Functional Annotations session together with Paul Thomas. So please submit your talks and papers! Papers will be reviewed separately from talks for a special issue of DATABASE.
Here’s the official announcement:
The International Biocuration Conference is a unique event for curators and developers of biological databases to discuss their work, promote collaborations, and foster a sense of community in this very active and growing area of research. For the 7th International Biocuration Conference in Toronto, Canada you are invited to submit your work for publication. This call for papers is done in collaboration with DATABASE: The Journal of Biological Databases and Curation. The DATABASE journal will publish an online Virtual Issue of the accepted papers. This is a great occasion to enhance the recognition of your work and of our profession by the greater biological research communities.
This year there are five topic sessions from which submitters are invited to select:
1. Clinical Annotations
2. Systems Biology Curation
3. Functional Annotations
4. Microbial Informatics
5. Data Integration and Data Sharing
The manuscript review process will be expedited for these papers and we will thus need to be firm on the submission deadline:
– Submission deadline: November 15, 2013
– First decisions: December 6, 2013
– Deadline for revisions: January 10, 2014
– Final decisions: February 21, 2014
– Conference: April 6-9, 2014
Authors wishing to submit to DATABASE for the 2014 Biocuration Virtual issue should go to the DATABASE home page (http://database.
To be considered for a Biocuration 2014 presentation you MUST register and SUBMIT an abstract for the meeting. Meeting registration will be opening up in time for the Submission deadline. The selection of oral presentations at the conference is not associated with the publication and review of the DATABASE Virtual Issue.
The proceedings of the 2013 meeting, the Biocuration 2013 Virtual Issue, are online: http://www.
Kind regards,
-Biocuration 2014 Organizing Committee
Note: a repost of a 2010 post I published for Ada Lovelace day. Unfortunately, I am too busy these days to write a new one. “Ada Lovelace Day is celebrated today to “…raise the profile of women in science, technology, engineering and maths.”
So without further ado:
She is a ‘ministering angel’ without any exaggeration in these hospitals, and as her slender form glides quietly along each corridor, every poor fellow’s face softens with gratitude at the sight of her. When all the medical officers have retired for the night and silence and darkness have settled down upon those miles of prostrate sick, she may be observed alone, with a little lamp in her hand, making her solitary rounds
— the Times newspaper, 8 February 1855
It’s Ada Lovelace Day, and time to write about your favorite woman in science.
Florence Nightingale is best known for founding the profession of modern nursing. Today nursing is a skilled degree-earning profession, requiring extensive training, with professional rights and responsibilities. That was not the case less than 120 years ago, when normally only the military and religious orders offered semi-skilled assistance to physicians. Nightingale changed all that, and revolutionizing the way medicine is practiced. Historically known as the “Lady with the Lamp”, the angel of soldiers in the Crimean War, she ministered to the wounded not only with care and compassion, but with a newly-applied professionalism. This professional approach included keeping medical records and using them to improve health care.
Nightingale is less known for her managerial and statistical acumen, and her pivotal role in medical statistics. Nightingale kept meticulous notes of mortality rates at the Scutari hospital in Istanbul which declined dramatically during her administration. Upon her return to London, she compiled the records into a new polar diagram, known as Nightingale Rose Chart. The data is plotted by month in 30-degree wedges. Red represents deaths by injury, blue – death by disease, and black – death by other causes.

Note that this is not a pie-chart. The wedges are all in 30-degrees (so 12 wedges/months fill a circle) and the contribution of each cause of death is proportional to each wedge’s radius. Nightingale’s visualization of the role preventable diseases play in battlefield deaths made a very strong case to military authorities, Parliament and Queen Victoria to carry out her proposed hospital reforms. Specifically for adopting hospital sanitation practices and dramatically reducing death from preventable infectious diseases.
Here is an interesting critique of the Nightingale Rose Charts which is presented at Dynamic Diagrams. It appears that by placing the preventable diseases wedge-section in the outer section of the wedge, the blue received a proportionally larger area, an artifact of this radial plot. This does not detract at all from her achievements, and, as shown in the corrected charts, not even from her case for improving hospital sanitation to reduce preventable diseases as the leading cause of death, regardless of presentation format. (Pie charts are usually problematic).
You can read more about the Mathematical affiliation of Nightingale in this excerpt form the Newsletter of the Association for Women in Mathematics. One interesting factoid: she was the first woman to be nominated a fellow of the Royal Statistical Society.
After a series of tweets and a couple of Facebook posts about the problems of the Journal Impact Factor (JIF), I was approached by a colleague who asked me: “so why are you obsessed with this”? My answer was that it irks me that I have to use the JIF next to my publications in so many different reports (grant reports, university annual activities, proposals, etc.) since it is a bad metric to evaluate the merit of my papers, and as a scientist, I do not like using bad metrics.
I assume that many readers of my blog constitute the proverbial choir on which my preaching would be wasted. Specifically, those who understand what the Thomson-Reuters Journal Impact Factor is, and how became such a poorly-understood and overused and abused metric. However, for those who have no idea what I am talking about, or for those who are thinking “what is wrong with the Impact Factor”? this post would hopefully be informative, if not valuable. It is a brief post. There was a lot written about the JIF, and the plausible alternatives that can be used to assess journal quality and impact, and I provide a list of further reading material sources at the end. Finally, for those who, like me, think that there are many wrong things with this metric and its use, and would like to convey that information, I hopefully provide some basic arguments.
What is the Journal Impact Factor?
The JIF is supposed to be a proxy for a journal’s impact: i.e. how much influence the journal has in the scientific community. It is calculated as follows:
A = number of times that articles published in the journal in years X-1 and X-2 from this journal were cited in year X
B = number of citable items in the journal in years X-1 and X-2
JIF(X) = A/B
Seems simple enough. The ratio of the number of citations to the number of publications. The higher the ratio, the more the articles are being cited. Therefore, the journal’s impact is higher.
Why is the JIF a bad metric? There are several reasons.

Continue reading Why not use the Journal Impact Factor: a Seven Point Primer →
As an ex-smoker I can attest to this: quitting is hard. It can also make you fat. I gained quite a few kilos when I quit, and those took a long time to lose. Happily, these days I am spending money on running marathons rather than on cigarettes.

Got a light?
Weight gain after smoking cessation is fairly well known. In fact, fear of weight gain is cited as a primary reason many smokers are reluctant to quit. There’s a large body of research on weight gain in those who quit. In fact, one thought is that smoking cessation, when accompanied by weight gain, can throw the quitter from Scylla to Charybdis: glucose tolerance becomes lower, and you are now in danger of developing metabolic disease if you do not watch your weight gain. Therefore, intervention against weight gain is common practice in smoking cessation programs.
But why do smokers gain weight? Common answers are: nicotine is an appetite suppressant (and we even may know how); the metabolism of smokers is higher. Also, food is used as a replacement for smoking: quitters need that dopamine surge they are not getting anymore from nicotine. They are getting it from food, and especially the fatty, salty and sweet food that is so unhealthy, yet (like cigarettes) is designed to be addictive.

mmmm……….
But now, a group in Switzerland has found something new that may affect in post-cessation weight-gain: gut bacteria. Apparently, changes to the diversity and composition of gut bacteria are profound, and those changes seem to affect weight gain. Reminder: the bacterial composition of the gut flora of obese and lean people are markedly different. In a series of observations in humans, and experiments in mice, gut flora have been shown to directly affect weight gain. The type of bacteria that predominate in the gut of obese mice (and people) break down sugar more efficiently. Hence, a vicious cycle is created where an obese person would gain more calories from the same food than a lean person. Fortunately, the cycle can be broken through change of diet. And, as shown more recently, possibly by fecal transplant.
(Personally, I’ll stick with the change of diet, thankyouverymuchindeed.)
But back to those who quite smoking (yay!) but started gaining weight as a result (grrrr…). The changes in the phyla of their gut bacteria resemble those of obese people, although with some important differences, which may or may not mean that the gut flora changes are responsible for weight gain. For instance, the species in the smokers’ pre-quitting gut flora was fairly similar to that of a lean person, but much less diverse. That is an interesting finding by itself: somehow, smoking reduces gut flora diversity; but why that is, and what kind of effect this has on smokers’ health, is the subject for other studies. The post-quitting flora, on the other hand, was mostly similar to that of an obese person, with the difference that the flora was more diverse than the pre-quitting flora. taken together, these findings are pretty incredible: smoking causes such profound changes to our body, that the number and type of bacteria that settle in our guts are different between smokers and non smokers. And if you are a smoker, less bacteria “like” you: smoking somehow selects for certain types of bacteria. To me, the main finding was the increase in microbial diversity after quitting, not the gain of Firmicutes which may or may not contribute to post-cessation weight gain.
So: quit smoking. Even your bacteria will thank you. Eventually, a diverse gut is a healthy one.
Biedermann et al. Smoking Cessation Induces Profound Changes in the Composition of the Intestinal Microbiota in Humans (2013) PLoS ONE 8(3): e59260. doi:10.1371/journal.pone.0059260
The Kavraki group at Rice University is looking to hire an enthusiastic postdoctoral researcher to enhance their research efforts in computational structural biology, drug design, and computational bioengineering. The group has significant expertise in the development of methods for motion planning for complex systems in robotics. Their Open Motion Planning Library (OMPL, http://ompl.kavrakilab.org) is now used both in academia and industry and is part of ROS (http://www.ros.org). Inspired by its successes in motion planning, the group is developing modeling frameworks and algorithms for analyzing large-scale conformational changes in molecules and molecular assemblies. Applications range from drug design to the decipherment of the inner mechanisms of viruses. The postdoctoral research associate will assist in these efforts. The position becomes available in January of 2014. The duration of the position is one year and can be renewed for a second year. Compensation is commensurate with experience.
Qualifications and Application
The ideal candidate will have a strong enthusiasm for interdisciplinary work. Required skills include excellent analytical skills, excellent software engineering skills, experience with programming in C++, and extensive exposure to biochemistry and biophysics. A Ph.D. in a related field is required for the position. Experience with Rosetta is highly desirable. In the past successful candidates have had degrees in Computer Science, Bioengineering, Biophysics, Physics, and Chemistry. Excellent communication and collaboration skills are required as the selected candidate will be expected to work closely with current lab members and with collaborators. To indicate interest please send a CV to Lydia Kavraki (kavraki@rice.edu). Applications will be accepted until the position is filled.
Environment
The Kavraki Group is a small but vibrant group and provides a stimulating working environment. Most of its alumni are now in academic or upper-level research positions. For more information about the group check http://www.kavrakilab.org. The Computer Science and Bioengineering Departments at Rice are part of the Gulf Coast Consortia for for Quantitative Biomedical Sciences and offer an unparalleled training environment (http://www.gulfcoastconsortia.org/home.aspx). Rice University is a leading research university in the vibrant urban setting of Houston, TX, the fourth largest city in the U.S. Rice is a small, private university with exceptional strengths in engineering, biomedicine, and nanotechnology. The Computer Science Department is among the top 20 in United States, while the Bioengineering Department is ranked among the top 10 departments in United States by US News & World Report.
WikiProject Computational Biology/ISCB competition announcement 2013
The International Society for Computational Biology (ISCB) announces an international competition to improve the coverage on Wikipedia of any aspect of computational biology. A key component of the ISCB’s mission to further the scientific understanding of living systems through computation is to communicate this knowledge to the public at large. Wikipedia has become an important way to communicate all types of science to the public. The ISCB aims to further its mission by increasing the quality of Wikipedia articles about computational biology, and by improving accessibility to this information via Wikipedia. The competition is open to students and trainees at any level either as individuals or as groups.
The prizes provided by ISCB for the best articles submitted will be:
1st prize – $500 (USD) and 1 year membership to the ISCB.
2nd prize – $250 (USD) and 1 year membership to the ISCB.
3rd prize – $150 (USD) and 1 year membership to the ISCB.
For more information go to http://en.wikipedia.org/wiki/Wikipedia:WikiProject_Computational_Biology/ISCB_competition_announcement_2013
Contact Person: Alex Bateman (agb@ebi.ac.uk)
Yesterday I received an email from Kristjan Liiva, a student at RWTH Aachen University Germany. Kristjan has developed a really cool dashboard to analyze and visualize the development of collaborative OSS projects by mining their mailing lists and software repositories. (If the link doesn’t work, try again later; the project is heavily under development). The result is a very interesting picture of social trends in collaborative OSS projects.
Kristjan has mined the mailing lists and repositories of Biopython, Bioperl and Biojava, all three bio* (‘biostar’) projects have large developer and user communities, and have been around for over a decade.
One thing Kristjan did, is create a graph for each year, where the nodes are people, and the edges are based on email communications. You see a map of what biopython looked like in the early days, 2000:
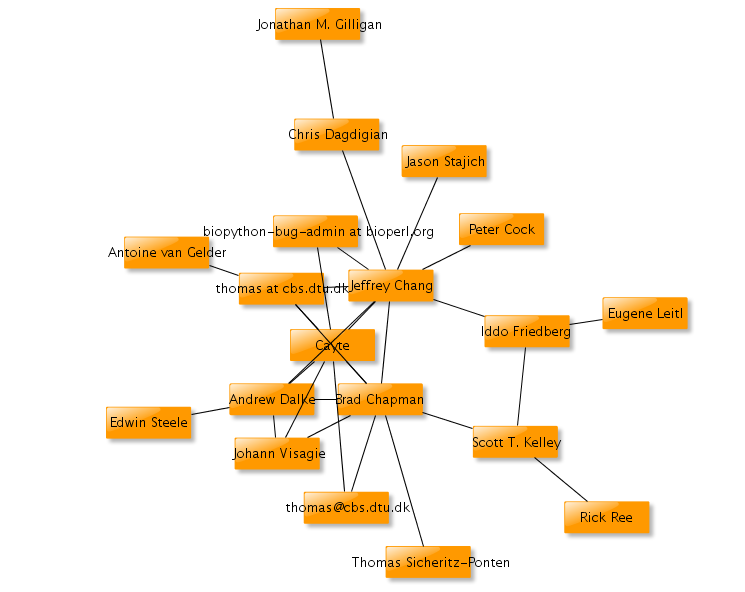
Note Jeffrey Chang (then a graduate student at Stanford), Andrew Dalke and Brad Chapman (then a graduate student at University of Georgia, Athens) with >5 edges each. They were quite busy at the time.
Continue reading The Bio* projects: a history in graphs →
Dear readers (Yeah, I’m talking to both of you!)
If you are a school teacher, college professor or any kind of other educator, trainer or science writer, and if you have ever used this blog in your line of work, please let me know. Also, if you are a student and used this blog as a source, please let me know. I would like to assess the impact of this blog for several reasons, including grant proposals and for a general assessment of the outreach and community impact of my department.
So: if you have used this blog in any way in your line of work (reading this blog to avoid work does not count, unfortunately), please tweet me (@iddux), or email me: idoerg ‘at’ gmail ‘dot’ com or just comment on this post. Please include:
Name
Profession / employer
How many posts did you use (roughly) and for what purpose? For example: “high school teaching, 3 posts”.

Announcing CAFA 2: The Second Critical Assessment of protein Function Annotations
Friends and Colleagues,
We are pleased to announce the Second Critical Assessment of protein Function Annotation (CAFA) challenge. In CAFA 2, we would like to evaluate the performance of protein function prediction tools/methods (in old and new scenarios) and also expand the challenge to include prediction of human phenotypes associated with genes and gene products. As the last time, CAFA will be a part of the Automated Function Prediction Special Interest Group (AFP-SIG) meeting that will be held alongside the ISMB conference. AFP-SIG will be held as a two-day meeting in July 2014 in Boston.
The targets and all information about the CAFA challenge are now available at http://biofunctionprediction.org. The submission deadline for predictions is January 15, 2014. The initial evaluation will be done during the AFP-SIG meeting in Boston. Anyone in the world is welcome to participate.
The mission of the Automated Function Prediction Special Interest Group (AFP-SIG) is to bring together computational biologists who are dealing with the important problem of gene and gene product function prediction, to share ideas and create collaborations. We also aim to facilitate interactions with experimental biologists and biocurators. We hope that AFP-SIG serves an important role in stimulating research in annotation of biological macromolecules, but also related fields.



