
Predicting protein function: what’s new?
![]()
(New: the paper was recently published in Genome Biology!)
Long time readers of this blog (hi mom!) know that I am working with many other people in an effort called CAFA: the Critical Assessment of protein Function Annotation. This is a challenge that many research groups participate in, and its goal is to determine how well we can predict a protein function from its amino-acid sequence. The first CAFA challenge (CAFA1) was held in 2010-2011 and published in 2013. We learned several things from CAFA1. First, that the top ranking function prediction programs generally perform quite well in predicting the molecular function of the protein. For example, a good function predictor can identify if the protein is an enzyme, and what is the approximate biochemical reaction it performs. However, the programs are not so good in predicting the biological process the protein participates in: it would be more difficult to predict whether your protein is involved in cell-division, apoptosis, both, or neither. For proteins that can influence different phenotypes (pleiotropic), or have different functions in different tissues or organisms, the predictions may even be worse.

The Human PNPT1 gene has several domains involved in several functions. Some CAFA1 methods (circled letters, each circle is a different method) predicted some of the functions. But only method (J) predicted two specific functions (3′-5′ exoribonuclease activity and Polyribonucleotididyltransferase activity). Some methods predicted “Protein binding” and “Catalytic activity” correctly, but those are very non-specific functions. Reproduced from Radivojac et al. (2013) under CC-BY-NC.
Another thing we learned from CAFA1 is that experimentally verified annotations in databases can be hugely biased because of high-throughput experiments that feed lots of data in the databases. In fact, we wrote a whole paper on that, The gist of it is in the following quote from another post:
Trouble is, once we started looking at the experimental annotations, we discovered that the annotating terms were hugely biased. A fifth of the experimentally-validated proteins in SwissProt were tagged only with the Gene Ontology term “Protein Binding” — a rather uninformative term. Yes, 20%! This prompted me to write the following protein function prediction program. Trust me, it works great, with a 20% recall value:
def predict_function: print("Protein Binding")The great thing about this program, is that it doesn’t even need input…
Anyhow, in CAFA2 we learned and improved; we also expanded in more ways than one. First, CAFA2 nearly doubled the number of teams and participants. We have so many co-authors on the latest manuscript (147), that I wrote a script just to manage the authors and their affiliations. Second, we provided many more targets on which to predict: 48,000 proteins in CAFA1, and 100,000 proteins in CAFA2. Third, we asked predictors to predict on more aspects of the function: in CAFA1 we only used two functional aspects: the molecular function and the biological process (from the Gene Ontology or GO). In CAFA2 we asked predictors to also predict the cellular component of the protein using GO terms and, for sequences from humans, also try the Human Phenotype Ontology (HPO). The HPO deals mostly with human disease phenotypes.
Fifth, we used two different metrics to measure how well methods are performing. In CAFA1 we only used precision and recall. Using precision/recall, a method that preforms well will have a combination of a high number of true positive predictions out of all predictions (high precision) and a high number of positive predictions out of all those predicted to be true (high recall). In CAFA2 we also used a measure we called “Semantic Distance“, developed by Predrag Radivojac and his (then) student Wyatt Clark that takes into account how informative a term is. Simply put, if a program correctly predicts terms that are more specific (say “Protein Tyrosine kinase activity”) than others (“Kinase activity”), that program will score higher. Semantic Distance also weighs the predicted terms by how much information they carry, so just predicting “Protein binding” (20% chance of success, remember?) will not get you far using Semantic Distance.
Sixth, we worked with biocurators, who provided us with some target proteins. Biocurators are the people who manually annotate proteins in databases . The databases that are monitored and populated mostly by biocurators (like SwissProt) are considered to be of much higher quality. For CAFA2, we had a team of biocurators lead by Rachael Huntley from EBI (now at University College London) with Maria Martin, Claire O’Donovan, and others provide us with many high quality annotations for human proteins that we used to challenge the participants in CAFA2.
There are many other interesting things: we assessed similarities between methods based on their predictions: which methods were most similar to each other? Why? We also looked at what are the functions that methods can best predict, and how well they can do that, we call it term-based prediction, as opposed to protein-centric prediction in which a a program is judged by how well it can assign a function given a certain protein).
Finally, to crown it all, we also ran a head-to-head analysis, pitting the top 5 methods in CAFA1 against the top 5 method in CAFA2.
Results
As in CAFA1, the top performing methods worked best in the molecular function aspect. Predictions of the biological process the protein participates in, or the cellular compartments it inhabits, were not as good, with biological process being the hardest to predict. The slideshow (click) below shows how well the top-10 methods performed, on a scoring scale (Fmax) of 0-1.


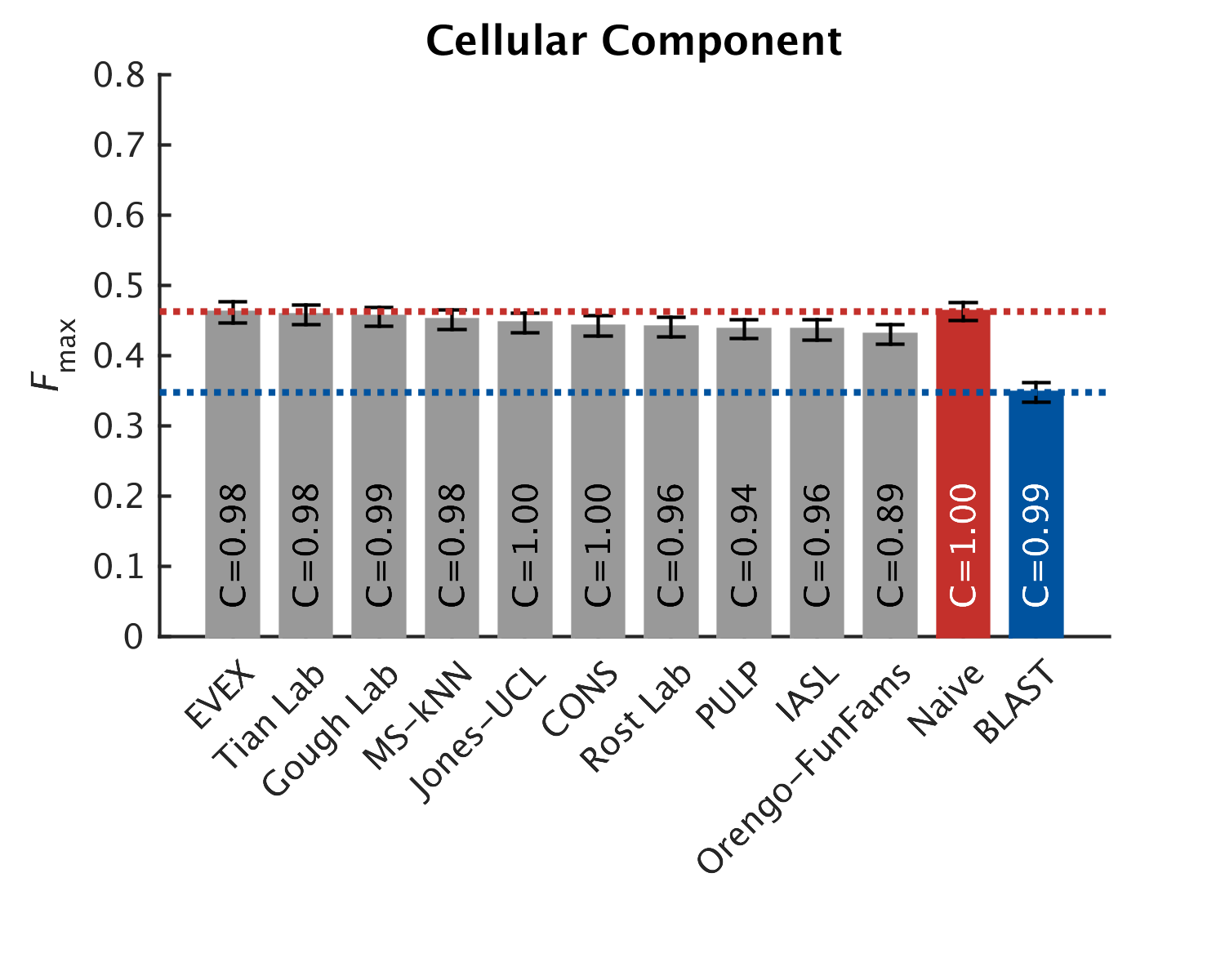

We also looked at how well methods predict for certain organisms. For example, here are the top methods that predict the function in mouse. As you can see, the performance here is more or less equivalent to that of the general predictions, mildly better for the Cellular Component ontology (click to activate the slideshow):
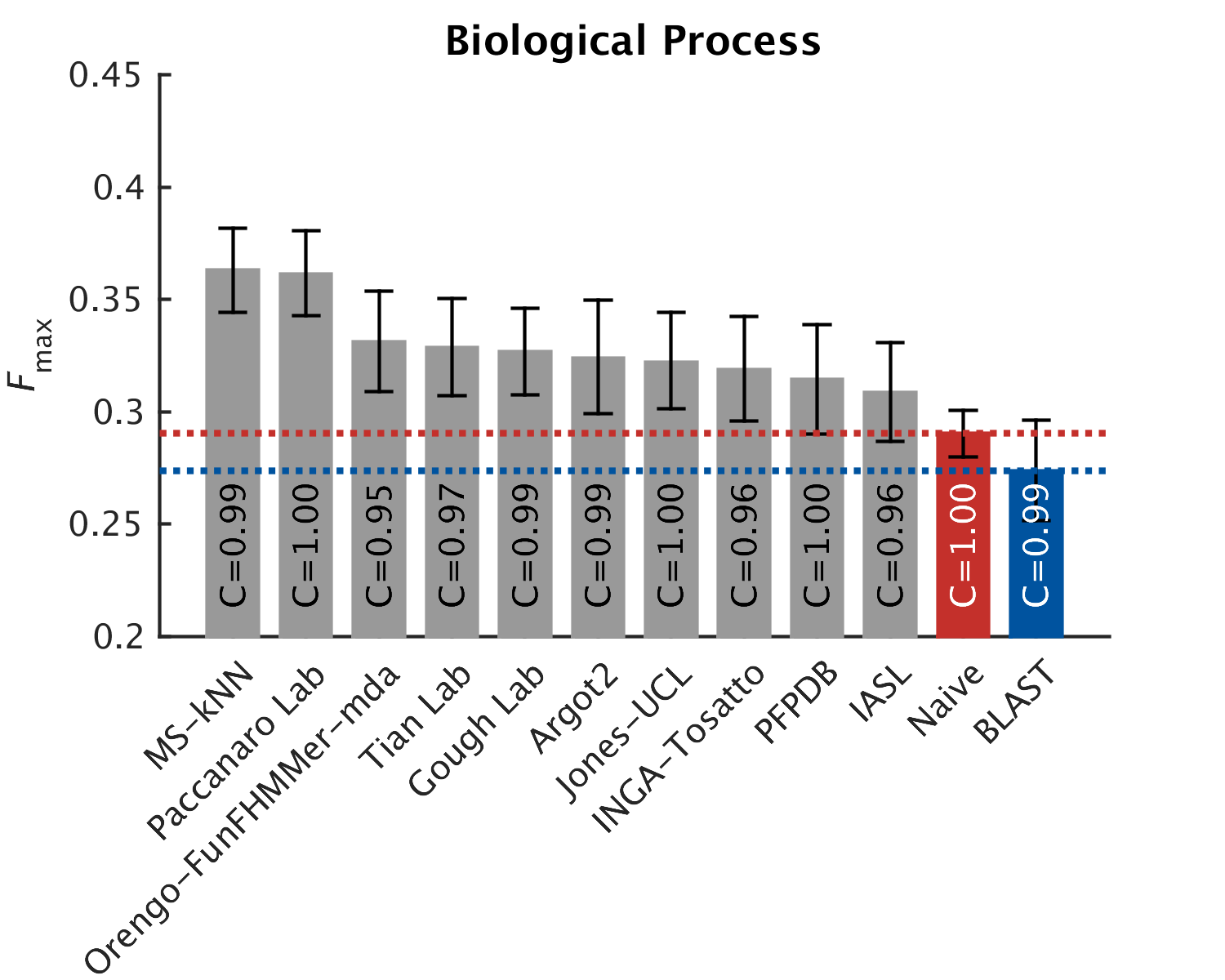
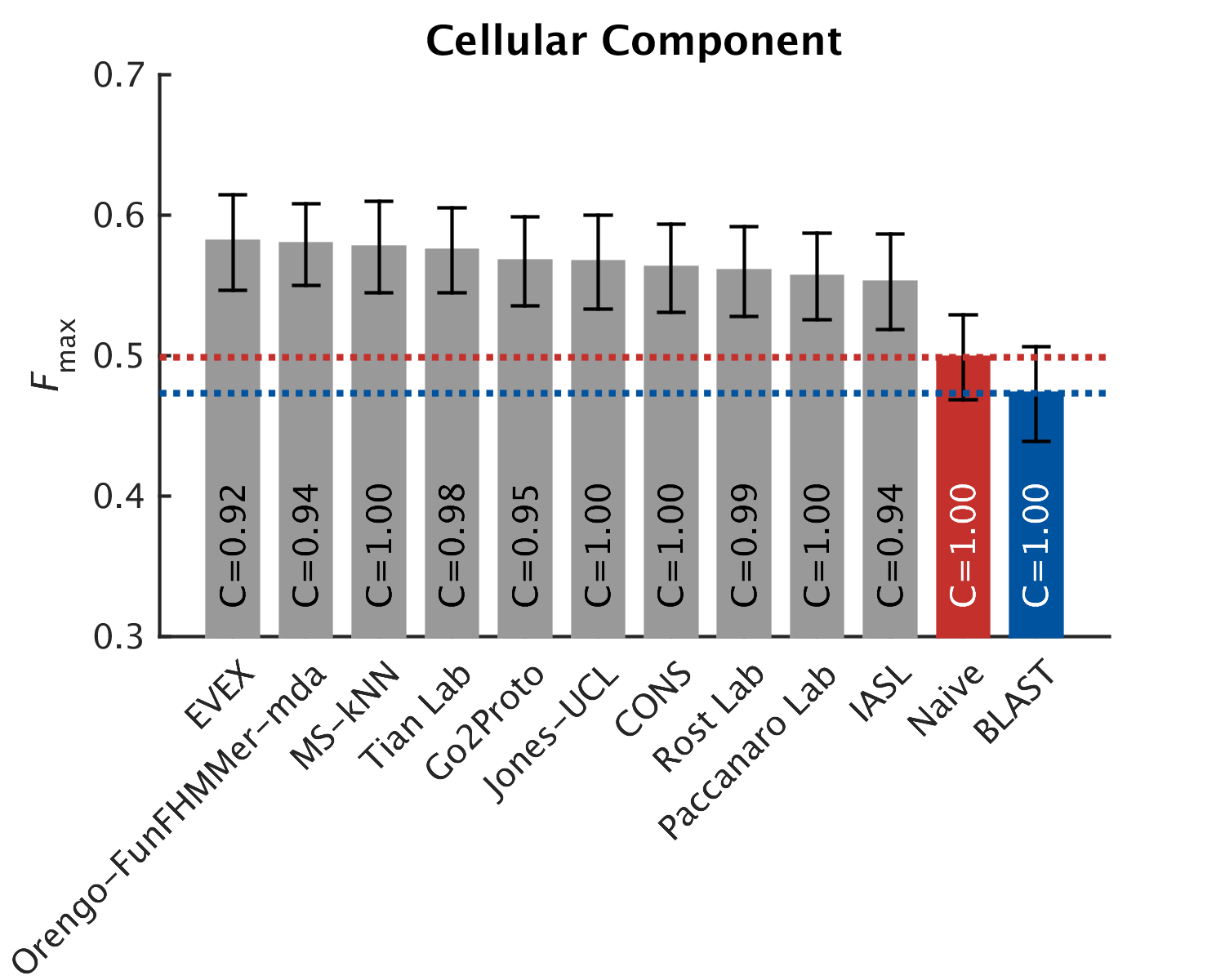
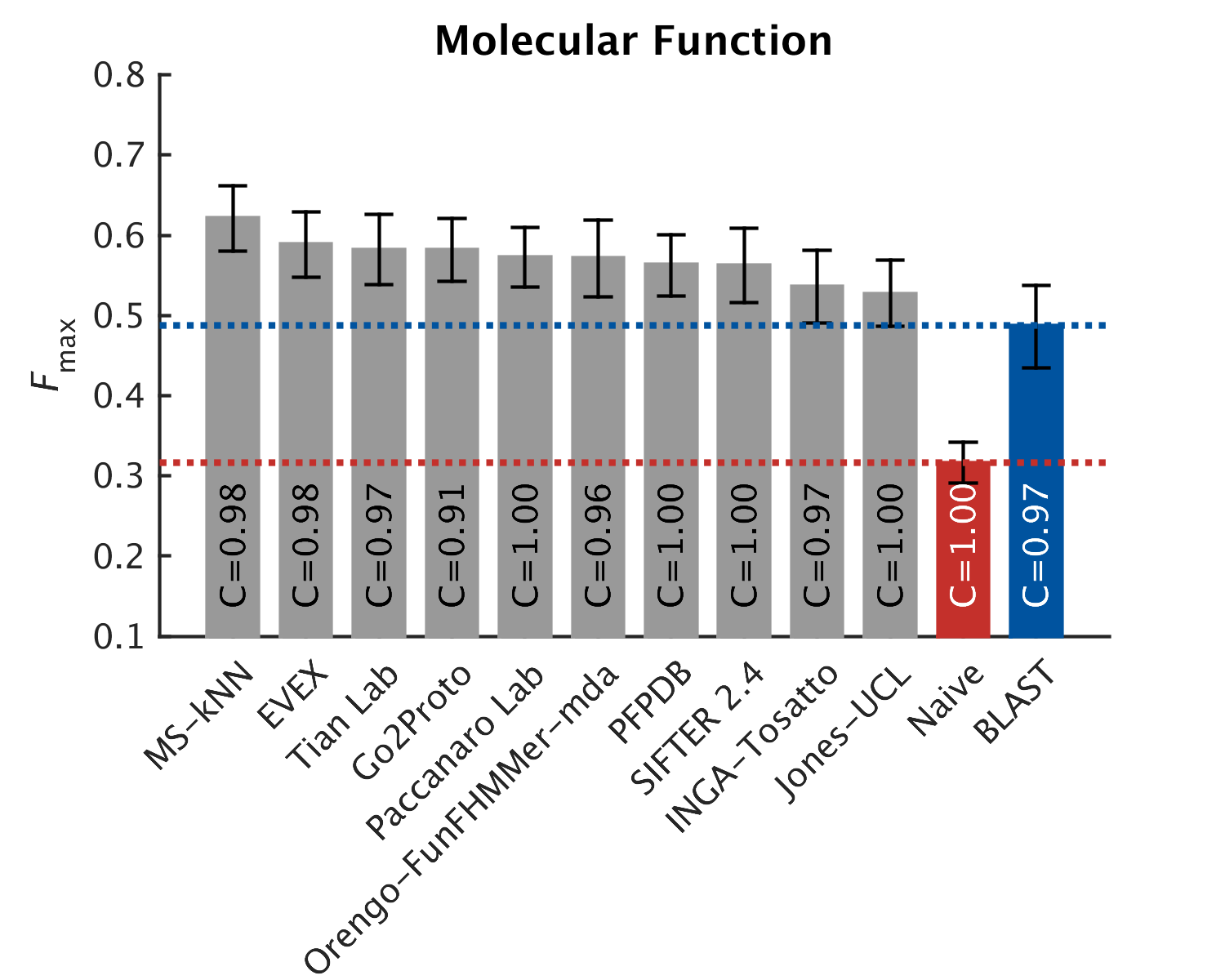
But good news! The top five methods in CAFA2 performed better than the top 5 of CAFA1, on a subset of proteins that is common to both challenges. This means that the ability to predict protein function is improving in the Biological Process ontology. We pitted the top 5 CAFA1 methods against the top 5 CAFA2 methods, and counted how many times CAFA2 won over CAFA1, the numbers are in the boxes below. The greener the box, the better CAFA2 methods performed. Not a single top CAFA1 method performed better than the top CAFA2 methods, which gives us hope that the field is moving forward. The predictions have especially improved in the weak suite: biological process predictions.
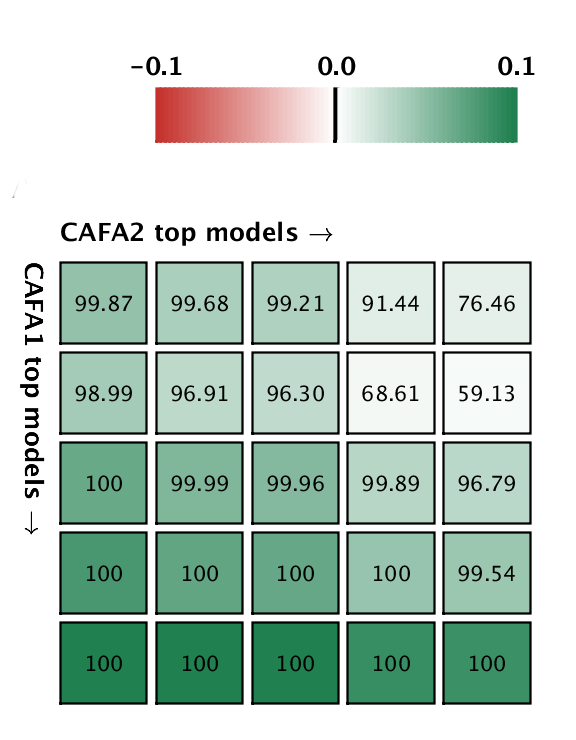
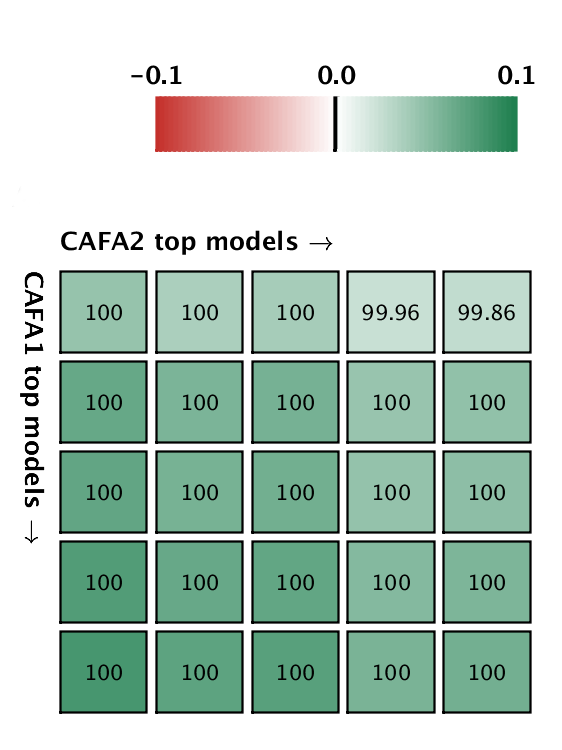
A comparison between the top 5 CAFA1 models against the top 5 CAFA2 models. Colored boxes encode the results such that (1) colors indicate margins of a CAFA2 method over a CAFA1 method, and (2) numbers in the box indicate the percentage of wins. Top: Molecular Function; bottom Biological Process.
More good news: we received funding from the National Science Foundation to continue with CAFA3, and to expand even more. CAFA3 will will start around September 2016 (fire up your computers), and will run through July 2017.
We produced tons of data from CAFA2, and anyone can delve into it and analyze. There are many interesting questions that can be asked: which methods seem to correlate better than others? Which methods perform best on which organisms and ontology combination? How is the structure of GO related to our ability to predict? When do the CAFA metrics best agree on the performance of the methods they assess and when don’t they?
Credits: the analysis work was mostly done in Predrag Radivojac’s lab, by his student Yuxiang Jiang who is also the lead author on the paper. There are many more people to thank: Sean Mooney and Tal Ronnen Oron made sure that the website ran smoothly and that people could easily submit their predictions. A programmer in my lab, Asma Bankapur, wrote some of the analysis scripts. Anna Tramontano and her group performed some of the analyses. Casey Greene from University of Pennsylvania contributed to the analysis as well. Our steering committee, Patricia Babbitt, Steven Brenner, Christine Orengo, Michal Linial and Burkhard Rost kept us on the straight and narrow, advising us through numerous phone calls. And, of course, all the participants in CAFA2, the scientists who are working hard to improve their methods help everyone make better discoveries in life science.
Some resources
- The Function Special Interest Group page
The CAFA2 manuscript preprint.- New September 8, 2016: The CAFA2 paper published in Genome Biology.
- All the CAFA2 data on FigShare
- Code used in the CAFA2 analysis.
- If you want to engage more, join our mailing list for news and discussions about protein function prediction, and CAFA.
Jiang Y, Oron TR, Clark WT, Bankapur AR, D’Andrea D, Lepore R, Funk CS, Kahanda I, Verspoor KM, Ben-Hur A, Koo da CE, Penfold-Brown D, Shasha D, Youngs N, Bonneau R, Lin A, Sahraeian SM, Martelli PL, Profiti G, Casadio R, Cao R, Zhong Z, Cheng J, Altenhoff A, Skunca N, Dessimoz C, Dogan T, Hakala K, Kaewphan S, Mehryary F, Salakoski T, Ginter F, Fang H, Smithers B, Oates M, Gough J, Törönen P, Koskinen P, Holm L, Chen CT, Hsu WL, Bryson K, Cozzetto D, Minneci F, Jones DT, Chapman S, Bkc D, Khan IK, Kihara D, Ofer D, Rappoport N, Stern A, Cibrian-Uhalte E, Denny P, Foulger RE, Hieta R, Legge D, Lovering RC, Magrane M, Melidoni AN, Mutowo-Meullenet P, Pichler K, Shypitsyna A, Li B, Zakeri P, ElShal S, Tranchevent LC, Das S, Dawson NL, Lee D, Lees JG, Sillitoe I, Bhat P, Nepusz T, Romero AE, Sasidharan R, Yang H, Paccanaro A, Gillis J, Sedeño-Cortés AE, Pavlidis P, Feng S, Cejuela JM, Goldberg T, Hamp T, Richter L, Salamov A, Gabaldon T, Marcet-Houben M, Supek F, Gong Q, Ning W, Zhou Y, Tian W, Falda M, Fontana P, Lavezzo E, Toppo S, Ferrari C, Giollo M, Piovesan D, Tosatto SC, Del Pozo A, Fernández JM, Maietta P, Valencia A, Tress ML, Benso A, Di Carlo S, Politano G, Savino A, Rehman HU, Re M, Mesiti M, Valentini G, Bargsten JW, van Dijk AD, Gemovic B, Glisic S, Perovic V, Veljkovic V, Veljkovic N, Almeida-E-Silva DC, Vencio RZ, Sharan M, Vogel J, Kansakar L, Zhang S, Vucetic S, Wang Z, Sternberg MJ, Wass MN, Huntley RP, Martin MJ, O’Donovan C, Robinson PN, Moreau Y, Tramontano A, Babbitt PC, Brenner SE, Linial M, Orengo CA, Rost B, Greene CS, Mooney SD, Friedberg I, & Radivojac P (2016). An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome biology, 17 (1) PMID: 27604469













Comments are closed.



