
The allure of the superficial
![]()
A new paper from my lab and Patsy Babbitt’s lab in UCSF has recently been published in PLoS Computational Biology. It is something of a cautionary tale for quantitative biologists, especially bioinformaticians and system biologists.
Genomics has ushered biology into the data rich sciences. Bioinformatics, developing alongside genomics, provided the tools necessary to decipher genomic data. But genomic data provides us with the instruction book: what the organism is capable of doing. To see what the organism actually does we need to run experiments to interrogate the biological pathways that together constitute life. But biochemical experiments only tell you about a few proteins at a time. Slowly. Much slower than the information gain from genomics.
A lab can spend decades deciphering a single biological pathway. A professor can spend her entire career investigating how a handful of proteins interact with each other and affect a certain cellular process. But in the age of genomics and systems biology this seems so dated; if we can sequence a human genome for $5,000 and take only a couple of days to do so, we would also like to analyze cellular pathways with the same ease. The rate by which we gain knowledge in genomics is much faster than that in molecular biology and biochemistry. And that can be frustrating.
But sequencing and pathway analysis are two different problems. The DNA sequence is immutable (I’m not talking about induced mutations or evolution over generations now), and the data contained therein is well-defined. Once you know the correct nucleotides (sequencing) and their order (assembly) , that is the sum total of the data. Of course, there is plenty more to do to analyze these data for gene location, splicing, etc, but the DNA sequence doesn’t change in the organism over time. Cellular networks, on the other hand, form and break up in response to the environment. Unlike a genome, they also vary from tissue to tissue and from cell to cell. So the information from cellular pathways is mutable. The components of a pathway perform, in many cases, in a probabilistic manner. Also, the information about each component of a pathway is ill-defined: the same protein may perform one function in pathway “A”, and in another way in pathway “B”. In terms of information content, cellular pathways are considerably richer, and the simple exercise in gathering data, unlike sequencing, is not a finite and well-defined task. Capturing pathway information is difficult, because it is transient, and ill-defined.
Enter systems biology with the promise of mitigating the knowledge gap, and the frustration that comes from it. High throughput assays such as Yeast 2 Hybrid, Mass Spectrometry proteomics, and RNAi functional genomics have accelerated the rate of data collection in cellular biology, and helped systems biology keep up with the Joneses — I mean genomics. Concurrently, computational tools were developed to analyze proteomes, interactomes, metabolomes and transcriptomes.
But how useful are data from high throughput experiments? My lab takes a leading part in the Critical Assessment of Function Annotation (CAFA) efforts, where we examine how well programs assign function to proteins. To create a good benchmark to test these software, we used proteins whose functional data came from experiments. Trouble is, once we started looking at the experimental annotations, we discovered that the annotating terms were hugely biased. 20% of the experimentally-validated proteins in SwissProt were tagged only with the Gene Ontology term “Protein Binding” — a rather uninformative term. Yes, 20%! This prompted me to write the following Protein function prediction program. Trust me, it works great, with a 20% recall value:
def predict_function:
print "Protein Binding"
The great thing about this program, is that it doesn’t even need input…
Anyhow, for CAFA we de-biased our benchmark by removing the proteins which had only the Protein Binding term as their annotation. But this led us to ask why this term became so dominant. A quick look at UniProt-GOA has shown us that only a few studies contributed this term. But these studies were of the high throughput variety, so they annotated many proteins. One we discovered that, we asked: “OK, so are there any other high throughput studies that annotate many proteins In fact, how much of what we know about proteins come from high throughput studies?” We discovered something rather shocking:
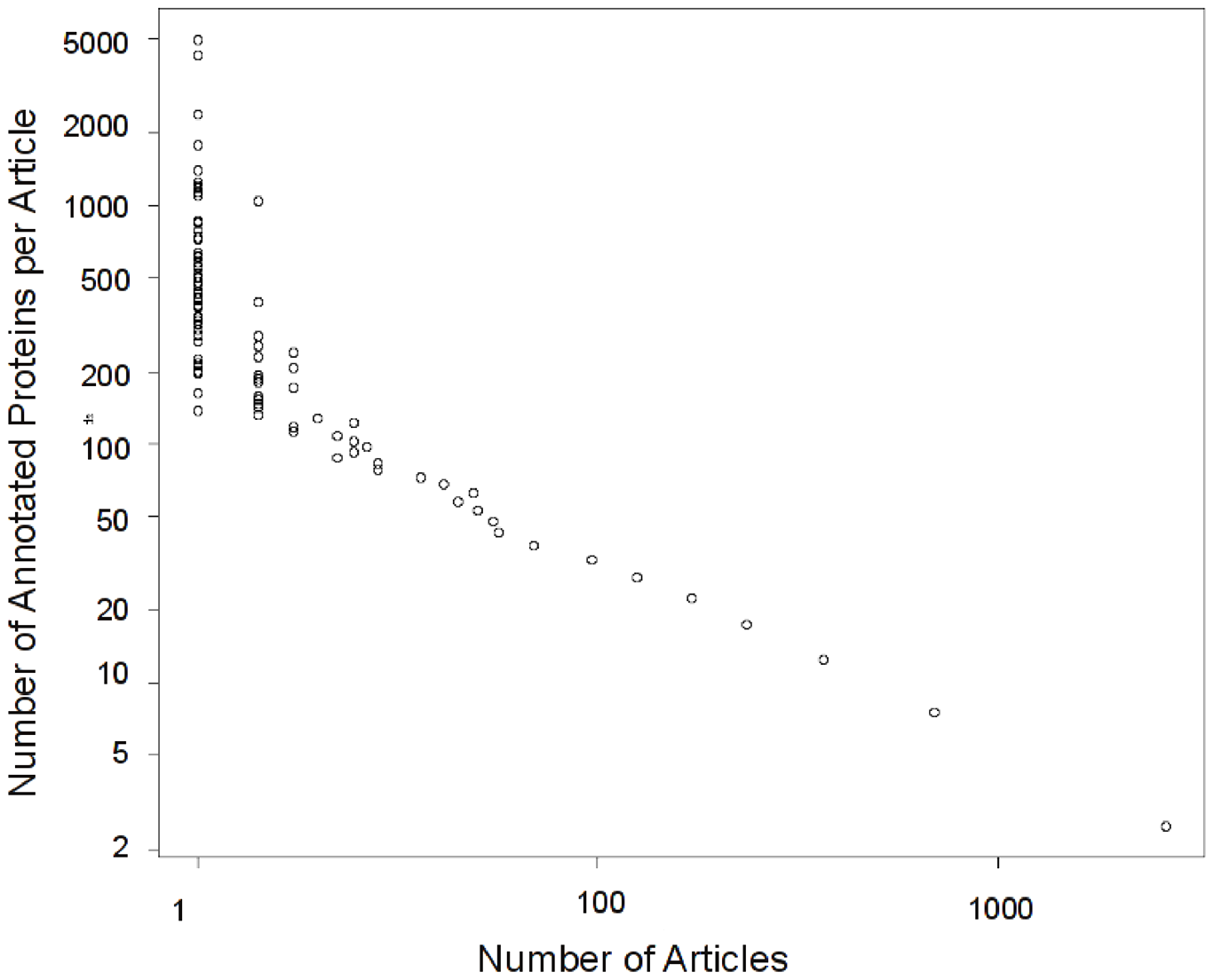
This distribution is logarithmic. There are very few studies that provide high throughput data, but those studies dominate what we know about protein function. Only 0.14% (zero point fourteen percent) of the studies provide 25% (yes!) of the experimentally validated protein annotations. Those are all high throughput studies: studies that provide experimental information on 100 or more proteins. Trouble is, as we discovered, the information that these studies provide has lower content. The information provided is mostly in very general terms “Protein Binding” , “Growth” or “Embryonic Development”. We can actually quantify how informative a term is, by looking at its frequency in the corpus, and its location in the GO term hierarchy. We found that the information content provided by high-throughput studies is considerably lower than that provided by low throughout studies. Here:
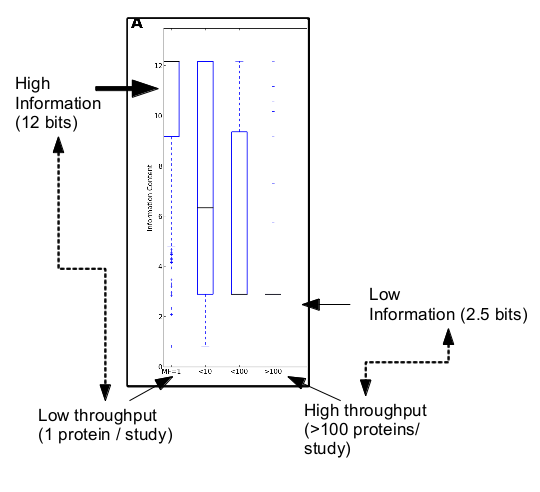 (For ontology geeks, that’s the information content for the Molecular Function Ontology in GO. We also looked at Biological Process and Cellular Component, the full results are in the paper.)
(For ontology geeks, that’s the information content for the Molecular Function Ontology in GO. We also looked at Biological Process and Cellular Component, the full results are in the paper.)
Paraphrasing Sydney Brenner, we have a ‘high throughput low output problem’. Functional annotations coming from ‘factory science’ carrying low information are taking over what we deem to know about protein function, masking out, by sheer mass, the higher quality annotations. Low information is somewhat worse than no information For bioinformaticians and other biologists doing large scale analyses it is tempting to use these data. But we should beware the allure of the superficial annotations. We need to understand that high throughput experiments can provide only so much information, and that information should be used very carefully when performing large-scale analyses. Otherwise, you might begin to think that Python two-line script is actually worth something.
This is a good opportunity to thank Alexandra Schnoes from Patsy Babbit’s lab, and Dave Ream and Alex Thorman from my lab for all their hard work. Also, the great folks at EBI’s UniProt-GOA team and especially Rachael Huntley for their work on UniProt-GOA, and for gently and patiently walking us through this invaluable resource.
Schnoes, A., Ream, D., Thorman, A., Babbitt, P., & Friedberg, I. (2013). Biases in the Experimental Annotations of Protein Function and Their Effect on Our Understanding of Protein Function Space PLoS Computational Biology, 9 (5) DOI: 10.1371/journal.pcbi.1003063

















Interesting; I guess not too surprising. (As someone from outside the field):
* Are the high throughput studies getting more sophisticated? – i.e. will the next generation of studies look for something even slightly more than ‘protein binding’; I’d expect to see a slow increase in information spread out across the whole organism as the techniques evolve.
* how useful are these types of annotations to the next guy along trying to do stuff manually?
Dave
You said:
“But sequencing and pathway analysis are two different problems. The DNA sequence is immutable (I’m not talking about induced mutations or evolution over generations now), and the data contained therein is well-defined. Once you know the correct nucleotides (sequencing) and their order (assembly) , that is the sum total of the data. Of course, there is plenty more to do to analyze these data for gene location, splicing, etc, but the DNA sequence doesn’t change in the organism over time. Cellular networks, on the other hand, form and break up in response to the environment. Unlike a genome, they also vary from tissue to tissue and from cell to cell. ”
Where do “transposable elements” fit into this apparently over-simplified description of the genome?
Transposable elements change a genome over generations. If I sequence your genome today, and then I sequence it again tomorrow, I will still get the same sequence.
I really agree with the differences between genome sequencing and other large scale experiments like protein-protein interaction mapping. Interactions are much more fuzzy and it will much harder to figure out when an interaction map is “complete”. Still, there are many benefits from doing large scale protein-protein, RNAi phenotyping, genetic-interactions screens etc. You get better cost per measurement and better benchmarking (usually) than focused single gene/protein measurements. What is not so easy to evaluate is how many specific studies follow up from large scale approaches. How much are the large scale efforts seeding more specific studies/. It would be hard to figure out just by tracking citations since a lot of people might bias their studies from something they saw in a database without ever citing the large-scale study or the database. It is clear that both types of studies (focused and large-scale) are needed but there is an important discussion to have on how much to allocate to each type.
> This prompted me to write the following Protein function prediction program. Trust me, it works great, with a 20% recall value:
Comment 1: ROTFL
Comment 2: I think you mean 20% precision. 😉
Comment 3: Awesome post and paper. Question: is there a straightforward way for me to find out whether annotations came from a high-throughput experiment?